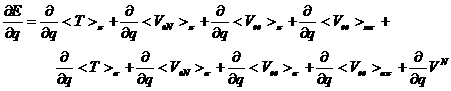|
Chem-Station-WhatsNew!-BBS-GuestBook-Chat-オークション-サイトマップ |
|
ベンゼン分子のπ電子は不安定な状態にある?2 |
|
ベンゼン分子のπ電子は不安定な状態にある?からの続きです。
▼ π電子のエネルギーとは?
共役したニ重結合のπ電子のエネルギーを計算するヒュッケル分子軌道法は,たぶんほとんどの有機化学者はこの方法を知っていると思いますので,計算方法の具体的説明は省略します.この分子軌道法は,Woodward-Hoffmann則の基にもなっています.昔から,ヒュッケル分子軌道法で得られたエネルギーをπ電子のエネルギーとよび,すでに述べたように,原子核,(固定した)s電子,および他のp電子が作るポテンシャルの中で運動するp電子のエネルギーであると理解されています.
ところで,CH2=CH-(CH=CH)n-CH=CH2 という系について調べるとπ電子のエネルギーには,nが1つ増えるごとに決まった量のエネルギーが増加するという不思議な性質があります.これをエネルギー加成性といいそれを基に芳香族性を定量的に定めています.なぜ不思議かというと,π電子のエネルギーであるにもかかわらず,原子核間の反発エネルギーも含んでいるからです.つまり,nが一つ増えることはCH=CHが加わることですが,これには2つの炭素原子核,2つの水素原子核も加わっています.
Ichikawaらは,この問題を調べるため全エネルギー(E)を5式とVNの和の形に分割し,ヒュッケル分子軌道によるp電子のエネルギーと比例関係にある項を探しました.その結果,全エネルギー(E),電子の運動エネルギー(
結論:π電子のエネルギーとはπ電子の運動エネルギー(<T>π)であり, EπShaikはπ電子のエネルギーには対応しない.
▼ πベンゼンで原子配置が変化したとき,π電子のエネルギーは比較出来るか
図5で,D6hベンゼン(B)をb2u座標上で結合距離をdだけ変化させたとします.(この意味は一つおきの結合をd伸ばしもう一つおきの結合をd縮めC構造のベンゼンにすることです.)この構造変化が,π電子のエネルギーに影響を与えなければ,構造B,C間の電子エネルギー変化はそのまま,π電子構造の変化に基づくことに起因するといえます.
表1. b2u 座標上でベンゼンを変形させたときのエネルギー変化a
term
benzeneb
distortedc
dif.d
E
-230.131181
-230.130948
0.6
Eel
-434.895341
-434.899241
-10.2
VN
204.764160
204.768293
10.9
<T>
229.990483
229.992533
5.4
<VeN>
-946.860745
-946.869539
-23.1
<Vee>
281.974922
281.977766
7.5
Epel
e
-40.067251
-40.068398
-3.0
EpShaik
f
-6.405036
-6.405731
-1.8
<T>p
7.471762
7.471929
0.4
<VeN>p
-85.535949
-85.537529
-4.1
<Vee>p
4.334722
4.334535
-0.5
Esel
g
-394.828090
-394.830843
-7.2
<T>s
222.518721
222.520603
4.9
<VeN>s
-861.324796
-861.332010
-18.9
<Vee>s
210.315770
210.317897
5.6
2X<Vee>ps
67.324430
67.325334
2.4
E'sh
-190.063930
-190.062550
3.6
E'sShaik
i)
-223.726145
-223.725217
2.4
aSTO-6G. bAu.
Geometry-optimized benzene (D6h). The bond lengths of C-C and C-H
are 1.38585 and 1.07867 Å. cAu. Displacement by 0.01Å along the b2u
coordinate (D3h). dDifference in kJ/mol. ep
Electronic energy. f‘p
Energy’ defined by Shaik. gs
Electronic energy. hSkeletal energy (E - Epel).
iSkeletal energy by Shaik.
d=0.01Åの場合,全エネルギーの変化はわずか0.6kJ/molです.一般にb2u座標上でのエネルギー変化は前に述べた理由により“わずか”になりますが,それでも分割したエネルギーの変化は大きくなり,最大で全エネルギー変化の39倍になっています.π電子のエネルギーに対する原子核配置の影響は<VeN>πに表れます.その値の変化(絶対値)をみると4.1kJ/molです.π電子のエネルギーの変化は,Shaikの定義では1.8kJ/molですので,Shaikらの議論でのエネルギー変化の主体は,π電子構造の変化に基づくのもではなく幾何構造変化によるということになります.
π電子構造とπ電子エネルギーの関係を求めようとしたはずなのに,実際はベンゼンの幾何構造とπ電子のエネルギーとの関係を調べたのです.
結論:幾何構造を変化させたとき起こるπ電子のエネルギー変化の大部分は原子核配置の変化によるものでπ電子構造の変化によるものではない.
▼
それなら,ベンゼンの問題はどのようにして解決するか
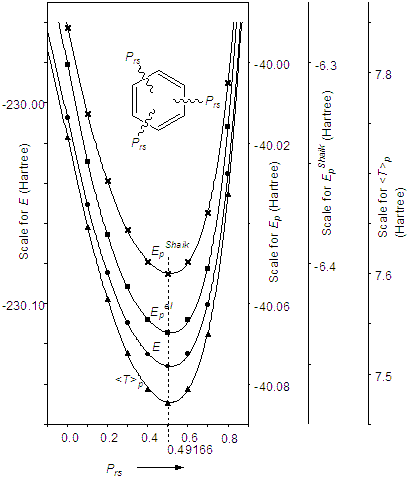
π電子のエネルギーはπ電子の運動エネルギー(<T>π)あることがわかりましたので,<T>πに注目すればよいことになります.幾何構造を変化させて電子のエネルギーを比較することはできないという問題に対して,Ichikawaらは2つの方法を用いて解決しています.一つは,幾何構造を変化させず,電子構造を変化させて全および分割エネルギーを電子構造の関数として得る方法です.それは,束縛条件付きHartree-Fock法とよばれます[33].もう一つは,エネルギーを核の配置あるいは結合次数で偏微分する方法(偏微分法)です[23].
▼ 束縛条件付きHartree-Fock法
電子構造を決めているのは波動関数です.波動関数は任意のものではなく,原子核や電子の電荷がつくる静電場[34]の中で定常波(定在波)である必要があります.定常波とは時間が経過しても存在する波でこれが重要なのです.原子軌道には主量子数,方位量子数,磁気量子数が現れますが,たとえば,主量子数が1.5などという原子軌道(波動関数)は,そのような波は存在出来ないから考えません.任意の電子構造に対応する定常波波動関数をどのようにして求めるかが問題となります.
の形で,λ がないものが通常のHartree-Fock方程式です.ここで,F,C,S,およびε
はそれぞれFock演算子,分子軌道への基底関数の寄与を表す係数,重なり積分および分子軌道のエネルギーです.λは系(分子)の電子構造を規定する演算子で,たとえば共役のないbenzeneの電子構造の波動関数を求めるなら,一つおきのC-C結合上のπ電子を掃き出す演算子と考えます.この方法の詳細は文献[33]に譲るとして,結果を紹介します.
図8.一つおきの結合次数(Prs)の関数として各種のエネルギーを表す
▼ 偏微分法による結果
エネルギー(E)を規定する変数をq(複数)として,それらのうち一つの変数qのみがΔq変化したとき,エネルギー(E)の変化(ΔE)の比を考えましょう.Δqを無限小のときの比をEのqに関する偏微分係数といいます.
qが方向を有しますので, も方向を持ちます.したがって,偏微分係数(値)は方向と大きさを持つ,ベクトル量となります.(なお,エネルギーは方向の情報を持たないスカラー量です.)
原子の核座標をqとすると,その位置での原子核に働く力となります.幾何構造が最適化されている場合は,すべての原子核座標の全エネルギーに関する偏微分係数が0となっています.また,分子の全エネルギーは原子核の座標の関数となっていますので,分子の(幾何)構造は,その近辺で小さな値となるようなエネルギーの「窪み」に入るような形で,原子核の位置がおさまります.このようなエネルギーの「窪み」は一般に複数あり,同じ分子式でいろいろな化合物が存在することに対応します.そのような点を,エネルギーの観点からは,エネルギーの極小値(local minimum)といいます.なお,エネルギーの最小値(global minimum)という言葉がありますが,これは複数ある極小値のうち最小のものをいいます.
さて,偏微分法を用いると,化学現象の原因を解明することができます.Eは5式とVNの和の形に分割できますので,6式は,
(12)
となり,qとして原子核座標をとれば,12式の右辺は原子核に働く力の成分となります.化学反応は原子核の位置の変化を伴いますので,12式の右辺を解析すればどのような種類の力がその反応を誘導しているかがわかります.12式のようなエネルギー分割成分の変数qに関する解析的微分法はTokiwaらにより与えられています[35]が,数値微分法でも容易に求めることができます.
ベンゼンはD6h構造のときEは最小値をとりますので,b2u座標上でbenzeneを変形させD3hとしそのときD6hに戻ろうとする力を解析すればbenzeneのD6h構造の原因が明らかになるはずです.b2u上で0.06A変形させた場合の値を表3に示します.
原子の核座標をqとすると,その位置での原子核に働く力となります.幾何構造が最適化されている場合は,すべての原子核座標の全エネルギーに関する偏微分係数が0となっています.また,分子の全エネルギーは原子核の座標の関数となっていますので,分子の(幾何)構造は,その近辺で小さな値となるようなエネルギーの「窪み」に入るような形で,原子核の位置がおさまります.このようなエネルギーの「窪み」は一般に複数あり,同じ分子式でいろいろな化合物が存在することに対応します.そのような点を,エネルギーの観点からは,エネルギーの極小値(local
minimum)といいます.なお,エネルギーの最小値(global
minimum)という言葉がありますが,これは複数ある極小値のうち最小のものをいいます.
ベンゼンはD6h構造のときEは最小値をとりますので,b2u座標上でbenzeneを変形させD3hとしそのときD6hに戻ろうとする力を解析すればbenzeneのD6h構造の原因が明らかになるはずです.b2u上で0.06A変形させた場合の値を表3に示します.
(2005.9.5 tanuki)
▼参考、関連文献
1.
Hiberty, P. C.; Shaik, S. S.; Lefour, J.-M.; Ohanessian, G. J.
Org. Chem. 1985, 50, 4657.
2.
Shaik, S. S.; Hiberty, P. C. J. Am. Chem. Soc. 1985,
107, 3089.
3.
Shaik, S. S.; Hiberty, P. C.; J.-M. Lefour, Ohanessian, G. J. Am.
Chem. Soc. 1987, 109, 363.
4.
Shaik, S. S.; Hiberty, P. C.; J.-M. Lefour, Ohanessian, G. Nouv.
J. Chim. 1985, 9, 385.
5.
Hiberty, P. C. Topics Curr. Chem. 1990, 153, 28.
6.
Hiberty, P. C.; Shaik, S. D.; Ohanessian, G.; Lefour, J.-M. J.
Org. Chem. 1986, 51, 3909.
7.
Hiberty, P. C.; Danovich, D.; Shurki, A.; Shaik, S. J. Am. Chem.
Soc. 1995, 117, 7760.
8.
芳香族性は有機化学の基本的概念ですので,どの有機化学の本にも載っています.ここでは,専門書いくつか挙げましょう.
a:“新しい芳香族系の化学”化学総説
15 日本化学会
編
b:
Badger, “Aromatic Character and Aromaticity,” Cambridge University Press,
London, 1969; Bergmann and Pullman, “Aromaticity, Pseudo-Aromaticity, and
Anti-Aromaticity, “Israel Academy of Sciences and Humanities, Jerusalem,
1971.
c: Minkin, Glukhovtsev, and Simkin, “Aromaticity and Antiaromaticity,”
A Wiley-Interscience,
9.
Hückel, E. Z. Phys. 1931, 70, 204.
廣田 穰
著
”分子軌道法”化学新シリーズ ,
裳華房
ISBN4-7853-3207-7.
10.
Kataoka, M.; Nakajima, T. J. Org. Chem. 1987, 52,
2323.
11.
Nakajima, T.; Kataoka, M.; Theoret. Chim. Acta 1992,
84, 27.
12.
Epiotis, N. D. Nouv. J. Chim. 1984, 8, 11.
13.
Cooper, D. L.; Gerratt, J.; Raimondi, M. Nature 1986,
323, 699.
14.
Heilbronner, E. J. Chem. Education, 1989, 66,
471.
15.
Jug, K; Koster, A. M. J. Am. Chem. Soc. 1990, 112,
6772.
16.
Janoschek, R. J. Mol. Struct. (Theochem), 1991, 229,
197.
17.
Baird, N. C. J. Org. Chem. 1986, 51, 3908.
18.
Aihara, J. Bull. Chem. Soc. Jpn. 1990, 63, 1956.
19.
Kollmar, H. J. Am. Chem. Soc. 1979, 101, 4832.
20.
Glendening, E. D.; Rüdinger, F.; Streitwieser, A.; Vollhardt, K. P.
C.; Winhold, F. J. Am. Chem. Soc. 1993, 115, 10952.
21.
Ichikaw, H.; Kagawa, H.; J. Phys. Chem. 1995, 99,
2307.
22.
Ichikaw, H.; Kagawa, H.; Bull. Chem. Soc. Jpn. 1997,
70, 61.
23.
Ichikaw, H.; Kagawa, H.; Bull. Chem. Soc. Jpn. 1997,
70, 727
24.
Ichikaw, H.; Kagawa, H.; Bull. Chem. Soc. Jpn. 1997,
70, 1805
25.
Ichikawa, H.; Yoshida, A.; Kagawa, H.; Aihara, J.; Bull. Chem.Soc.
Jpn. 1999, 72, 1737.
26.
Hornig, D. F. J. Am. Chem. Soc. 1950, 72, 5772.
27.
Berry, R. S. J. Chem. Phys. 1961, 35, 2253.
28.
全エネルギーをその成分の和の形に表す方法をエネルギー分割法とよばれ,比較的昔から用いられている手法です.詳しくは,下記の文献およびそこに引用されている文献を参照してください.Ichikawa,
H.; Yoshida, A.; Int. J. Quantum Chem., 1999, 71, 35. |
【用語ミニ解説】
■
■
■
■
|